Thalassemia là bệnh gì?
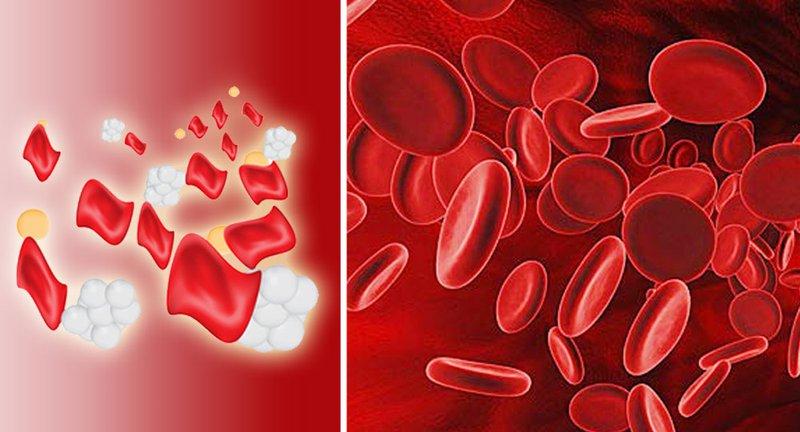
Nếu người bệnh tan máu bẩm sinh không được điều trị sớm sẽ xuất hiện nhiều biến chứng do thiếu máu và thừa sắt gây ra trên tất cả các cơ quan làm thay đổi diện mạo.
Bệnh tan máu bẩm sinh là một bệnh lý huyết học di truyền liên quan đến sự bất thường cấu trúc hemoglobin (một cấu trúc protein trong hồng cầu có chức năng vận chuyển oxy). Ở bệnh nhân Thalassemia, các chức năng của hồng cầu bị thay đổi dẫn đến tình trạng thiếu máu.
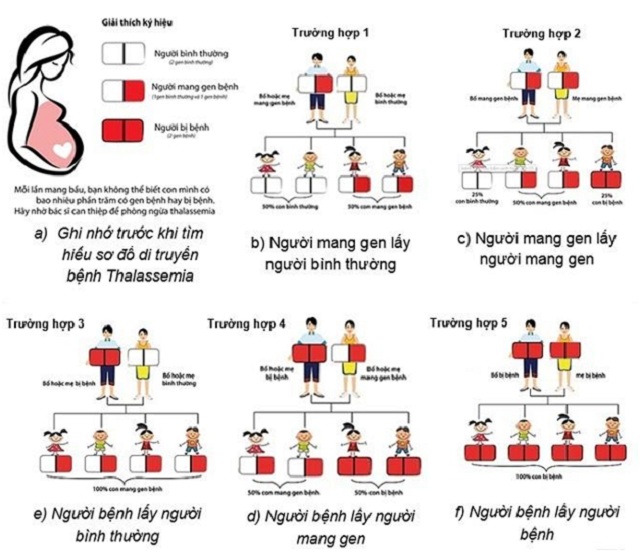
Thalasemia là một bệnh di truyền lặn trên nhiễm sắc thể thường. Cụ thể, người bình thường có 2 gen khỏe mạnh, người bị bệnh có 2 gen bệnh, người mang gen có 1 gen bệnh và 1 gen khỏe mạnh. Do đó, Thalassemia gây ra những hậu quả nghiêm trọng đến giống nòi, ảnh hưởng đến cuộc sống của bệnh nhân và cả cộng đồng.
Sự phát triển trong cộng đồng của Thalassemia
Thalassemia là một bệnh di truyền huyết học phổ biến nhất trên thế giới, trong đó Địa Trung Hải, Trung Đông, châu Á – Thái Bình Dương là những vùng có tỷ lệ mắc bệnh và mang gen bệnh cao. Bệnh Thalassemia xuất hiện ở cả nam và nữ. Tỷ lệ người mang gen bệnh là khoảng 7% dân số thế giới, trong đó có khoảng 1.1% cặp vợ chồng có nguy cơ sinh con bị bệnh hoặc mang gen bệnh Thalassemia, ước tính mỗi năm có khoảng 300.000 đến 500.000 trẻ sinh ra mắc Thalassemia ở mức độ nặng.
Tại Việt Nam, bệnh Thalassemia được ghi nhận từ năm 1960, hiện nay có khoảng 10 triệu người mang gen bệnh, khoảng 20.000 người bị Thalassemia thể nặng, ước tính mỗi năm có khoảng 2.000 trẻ sinh ra mắc bệnh Thalassemia. Bệnh phân bố khắp cả nước, phổ biến hơn ở các dân tộc ít người, các tỉnh miền núi, cao nguyên: tỷ lệ mang gen bệnh Thalassemia đối với dân tộc Mường là khoảng 22%, các dân tộc Êđê, Tày, Thái,… trên 40%, trong khi tỷ lệ này ở người Kinh khoảng 2 – 4%.
Nguyên nhân của bệnh Thalassemia

Bệnh nhân Thalassemia nếu không được điều trị có nguy cơ tử vong khi chỉ 15 - 20 tuổi.
Hemoglobin gồm có 2 thành phần là Hem và globin, trong globin gồm có các chuỗi polypeptid. Bệnh Thalassemia xảy ra khi có đột biến tại một hay nhiều gen liên quan đến sự tổng hợp các chuỗi globin, dẫn đến tình trạng thiếu hụt các chuỗi globin này, làm cho hồng cầu vỡ sớm (tan máu), và biểu hiện triệu chứng thiếu máu. Bệnh nhân mắc Thalassemia có thể nhận gen bệnh từ bố hoặc mẹ, hoặc cả bố và mẹ.
Bệnh được gọi tên theo chuỗi globin bị khiếm khuyết, gồm có 2 loại bệnh Thalassemia chính:
α-Thalassemia: Thiếu hụt tổng hợp chuỗi α, do đột biến tại một hay nhiều vị trí trên gen tổng hợp chuỗi α-globin.
β-Thalassemia: Thiếu hụt tổng hợp chuỗi β, do đột biến tại một hay nhiều vị trí trên gen tổng hợp chuỗi β-globin.
Triệu chứng bệnh Thalassemia
Bệnh nhân Thalassemia có thể vào viện với các dấu hiệu:
- Người mệt mỏi, hoa mắt chóng mặt.
- Da nhợt nhạt, xanh xao; có thể vàng da, vàng mắt.
- Nước tiểu vàng sẫm.
- Khó thở khi gắng sức.
- Trẻ chậm lớn.
Các biểu hiện của bệnh Thalassemia
Bệnh Thalassemia có 2 thể bệnh là α-Thalassemia và β-Thalassemia.
α-Thalassemia:
- Thể nhẹ (thể ẩn): thường không có triệu chứng, nếu có cũng chỉ gây thiếu máu nhẹ. Thường chỉ được phát hiện khi có các bệnh lý kèm theo hoặc vào các thời kỳ cơ thể tăng nhu cầu máu như mang thai, đa kinh,…
- Thể rối loạn Hemoglobin H: gây vàng da, gan lách lớn, dinh dưỡng kém. Có thể gây ra các biến dạng trên xương: má, trán, hàm có thể phát triển quá mức.
- Thể phù thai: Hầu hết trẻ mắc Thalassemia thể phù thai đều chết ngay khi sinh hoặc chết non
β-Thalassemia:
- Thể nhẹ: thường không có triệu chứng, nếu có cũng chỉ gây thiếu máu nhẹ.
- Thể trung gian: Biểu hiện với các triệu chứng tương tự như thể nặng nhưng ít trầm trọng hơn và tiến triển chậm hơn. Thường có biểu hiện thiếu máu rõ khi trẻ trên 6 tuổi, và lúc này mới cần phải truyền máu. Tuy nhiên, nếu không được điều trị, các biến chứng cũng có thể xảy ra ở bệnh nhân Thalassemia thể trung gian.
- Thể nặng: Thường xuất hiện sớm, có thể ngay sau khi sinh, hiếm khi xuất hiện sau 2 tuổi. Bệnh biểu hiện rõ nhất ở tháng thứ 4 – 6 với tình trạng thiếu máu trầm trọng và ngày càng nặng hơn, có thể đe dọa tính mạng. Có thể có tình trạng vàng da, mắt; lách to; thường xuyên bị nhiễm trùng. Thalassemia thể nặng đòi hỏi cần truyền máu nhiều lần.
Nếu được truyền máu đầy đủ, trẻ có thể phát triển bình thường cho đến 10 tuổi. Sau đó, có thể xuất hiện các biến chứng như:
- Biến dạng xương ở mặt, mũi tẹt, răng vẩu; loãng xương, dễ gãy xương.
- Da sạm, củng mạc mắt vàng.
- Sỏi mật.
- Dậy thì muộn ở nữ.
- Chậm phát triển thể chất.
- Biến chứng tim mạch: suy tim, rối loạn nhịp tim,…
Đường lây truyền bệnh Thalassemia
Bệnh Thalassemia không phải là bệnh truyền nhiễm như bệnh viêm gan virus, lao phổi,… Đây là bệnh di truyền do bệnh nhân nhận gen bệnh từ bố và mẹ. Do đó, bệnh Thalassemia là một bệnh có tính chất gia đình.
Đối tượng nguy cơ mắc bệnh Thalassemia
Những người sống trong các vùng dịch tễ của bệnh như Địa Trung Hải, Trung Đông, châu Á, châu Phi là các đối tượng có khả năng mắc bệnh Thalassemia cao.
Nguy cơ mắc bệnh sẽ tăng lên nếu trong gia đình có người bị Thalassemia hoặc có nguồn gốc tổ tiên từ châu Phi (đặc biệt là người Mỹ gốc Phi) và gốc Đông Nam Á, Địa Trung Hải.
Biện pháp phòng ngừa bệnh Thalassemia

Khám sức khỏe tiền hôn nhân và tiền mang thai giúp sàng lọc phát hiện bệnh sớm.
Tầm soát phát hiện bệnh sớm: Tư vấn tiền hôn nhân, đặc biệt là những người thuộc nhóm đối tượng nguy cơ của bệnh cần được tư vấn, làm các xét nghiệm để được phát hiện bệnh Thalassemia sớm. Nếu cả 2 vợ chồng đều mang một thể bệnh Thalassemia, nên được tư vấn trước khi có ý định mang thai.
Đối với các cặp vợ chồng cùng mang một thể bệnh Thalassemia, trong thai kỳ nên làm các xét nghiệm tầm soát và chẩn đoán gen đột biến khi thai được 12 – 18 tuần. Phương pháp được thực hiện có thể là chọc ối hoặc sinh thiết gai nhau để tìm đột biến gen (nếu có).
Các biện pháp điều trị bệnh
- Điều trị thiếu máu:
Chỉ định: Bệnh nhân có chỉ định truyền máu khi 2 lần kiểm tra đều cho thấy Hb<7g/dl, hay Hb>7g/dl nhưng có biến dạng xương.
- Truyền máu:
Bệnh nhân được truyền hồng cầu lắng, máu mới, lượng 10ml/kg trong 2-3h. Tần suất truyền máu có thể mỗi 4-6 tuần một lần tùy mức độ. Bệnh nhân cần được theo dõi chặt chẽ trong quá trình truyền máu tại bệnh viện.
Mục đích: duy trì Hb>10g/dl, giúp trẻ phát triển bình thường, tránh biến dạng xương.
- Điều trị ứ sắt:
Chỉ định thải sắt được đặt ra ở trẻ trên 3 tuổi, khi mà ferritin huyết thanh >1000ng/ml
Mục đích: ngăn ngừa tổn thương các cơ quan do ứ sắt, đặc biệt là tim và nội tiết.
Desferoxamin tiêm dưới da 30-50mg/kg trong 8-12h × 5-7 ngày/tuần, hoặc tiêm tĩnh mạch lúc truyền máu.
Chú ý theo dõi thị lực, thính lực hàng năm.
- Cắt lách:
Chỉ định: Thalassemia thể nặng, cường lách (lách to, giảm 3 dòng tế bào máu, lượng hồng cầu lắng truyền >250ml/kg/năm).
Biến chứng sau cắt lách: Nhiễm trùng, tắc mạch.
- Điều trị hỗ trợ: Vitamin C, Vitamin E, Acid folic.
- Ghép tủy xương: Là phương pháp hiện đại cho kết quả tốt trong điều trị Thalassemia. Tuy nhiên, ghép tủy xương có hạn chế là khó tìm được người cho tế bào gốc phù hợp.
Tin vui cho các cặp vợ chồng mang gen bệnh Thalassemia có nguy cơ cao sinh con mang bệnh và các bác sĩ hỗ trợ sinh sản là mới đây tạp chí Hỗ trợ Sinh sản và Di truyền (Journal of Assisted Reproduction and Genetics) của Hiệp hội Y học Sinh sản Hoa Kỳ (ASRM) đã đăng tải 1 nghiên cứu của Việt Nam về phương pháp sàng lọc phôi PGT-M-thalassemia mới. Nghiên cứu được thực hiện bởi nhóm tác giả đến từ Bệnh viện Phụ sản Hà Nội, Bệnh viện Nam học và Hiếm mộn Hà Nội và Công ty Cổ phần Dịch vụ Phân tích Di truyền (GENTIS).
GENTIS là đơn vị tiên phong về xét nghiệm gen tại Việt Nam, luôn chú trọng vào hoạt động nghiên cứu và phát triển, nhằm không ngừng cải tiến chất lượng các dịch vụ có sẵn song song với xây dựng và đưa ra các dịch vụ mới tiên phong để “ngày càng nâng cao chất lượng thể chất và trí tuệ người Việt”.
Bên cạnh cơ sở vật chất đầy đủ, hiện đại, sẵn sàng cho quá trình nghiên cứu cũng như quá trình cải tiến sản phẩm, GENTIS còn tự hào với đội ngũ nhân viên có trình độ cao, tay nghề thành thạo, có đam mê và hiểu biết những tiến bộ về công nghệ, kỹ thuật, liên tục cập nhật những kiến thức mới của ngành trên thế giới. Đặc biệt, GENTIS luôn có Hội đồng khoa học với các chuyên gia là những nhà nghiên cứu hàng đầu và rất giàu kinh nghiệm.
GENTIS đã và đang tập trung vào triển khai, nhận chuyển giao, nghiên cứu và phát triển các dòng xét nghiệm:
- Các xét nghiệm phân tích bệnh di truyền trên phôi tiền làm tổ (PGT-M): các xét nghiệm đã được đưa vào dịch vụ bao gồm PGT-M cho bệnh Alpha-Thalassemia, Beta-Thalassemia và Teo cơ tủy (SMA)
- Sàng lọc trước sinh không xâm lấn - NIPT: đã hoàn thành quá trình nhận chuyển giao công nghệ từ Illumina – trở thành đối tác đầu tiên và duy nhất cho đến nay của Illumina tại Việt Nam về xét nghiệm NIPT.
Sứ mệnh của GENTIS chính là không ngừng phát triển để trở thành đơn vị tiên phong trong lĩnh vực xét nghiệm gen, áp dụng những công nghệ xét nghiệm tiên tiến nhất để phục vụ cho khách hàng.